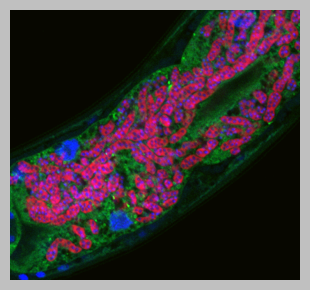
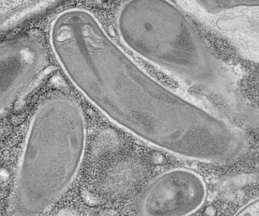
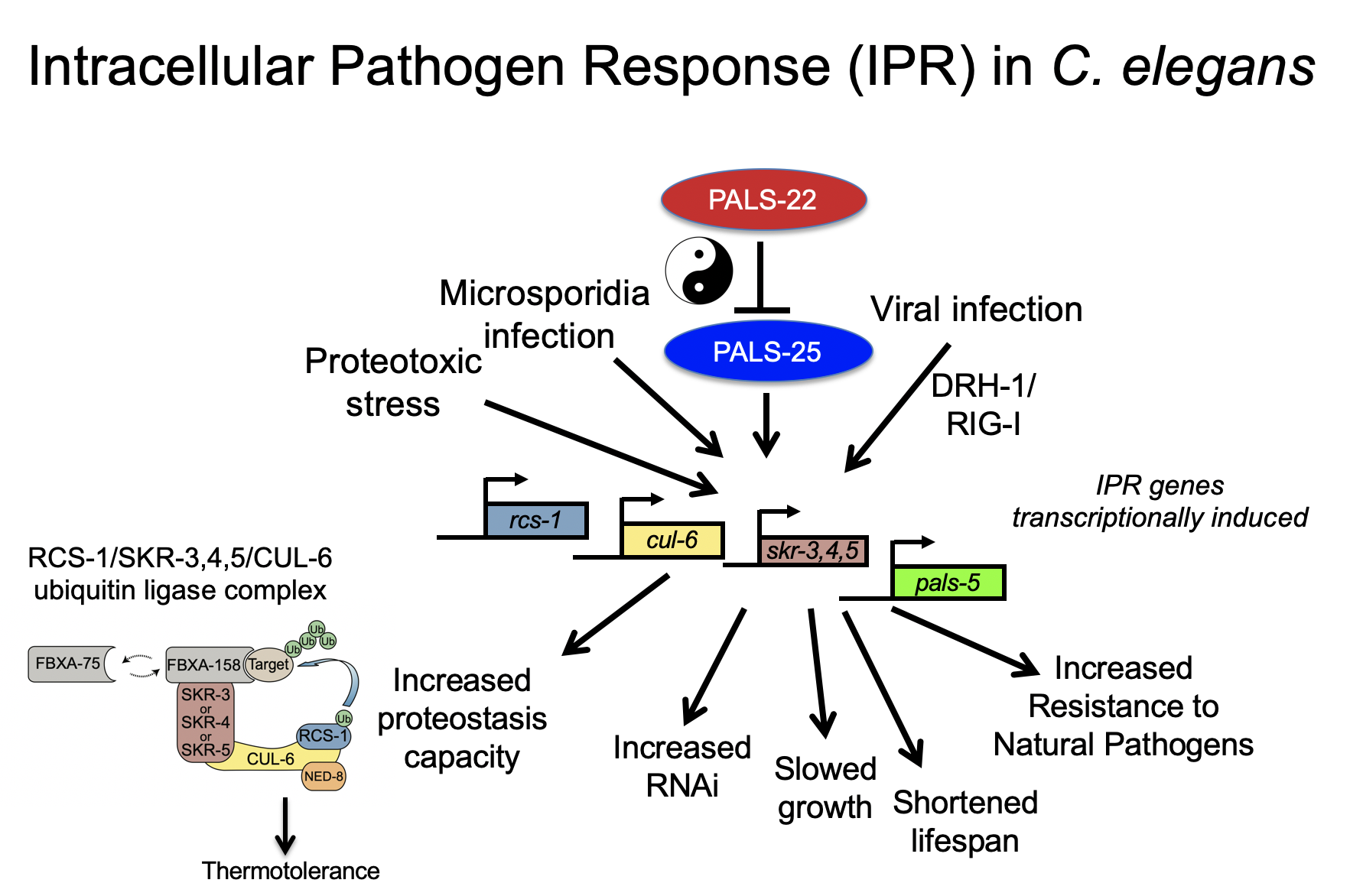
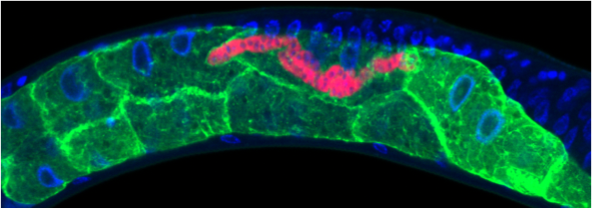
How did microsporidia evolve to be such successful pathogens? We have been involved in identifying new species of microsporidia that infect C. elegans (Luallen et al, 2016) to develop models to investigate this question. Most of our efforts focus on Nematocida parisii, which was the first pathogen shown to infect C. elegans in the wild (Troemel et al, 2008), and appears to be the most common species of microsporidia found in wild-caught C. elegans (Zhang et al, 2016). In previous work we have been involved in Microsporidia genome sequencing and transcriptomics to determine which gene gains and losses are specific to the Microsporidia phylum, in comparison to other fungi (Cuomo et al, 2012). We have also investigated the microsporidia ‘host-exposed’ proteome, to determine which factors microsporidia uses to manipulate host physiology (Reinke et al, 2017).